
【研究背景】
作為電動汽車和電網的下一代儲能技術,鈉電池展現出巨大的潛力,與鋰元素相比,鈉元素的天然丰度高、成本更低。鈉金屬負極具有較高的理論比容量(1165 mAh/g)和低氧化還原電位(2.714 V vs. SHE),將成為是鈉電池的終極負極材料。然而,傳統電解質與高反應性鈉金屬負極和正極的不相容性限制了其性能發揮,這些電極的平衡電位在傳統電解質的穩定電壓窗口之外,導致電解質易發生分解和電極形成不溶性產物。在正極側,界面副反應也會引起正極顆粒不可逆的表面相變和極化反應,導致容量衰減。在負極側,在鈉金屬沉積和剝離過程中,固體電解質界面(SEI)的化學-機械不穩定性導致形態降解和金屬分離,加速電解質消耗,降低電池可逆性和循環壽命。此外,枝晶的存在會橋接電極,導致內部短路和熱失控。最近,對鈉金屬電池(SMBs)的研究主要集中在金屬鈉的電沉積和剝離行為,以及SEI的結構和組成。為了減輕孤立Na0的積累並提高庫侖效率(CE),研究者提出了各種策略,包括電解質工程,界面功能化,多孔電極開發,和循環方案優化等。
對於電解質,開發了不同的鹽、溶劑、添加劑及其組合,目的是獲得更寬的氧化還原電壓和循環可逆性。單鹽單溶劑電解質需要在低極化和高氧化還原穩定性之間進行權衡。傳統電解質使用強陽離子溶劑化溶劑,增強陽離子-偶極相互作用,促進溶劑分離離子對(SSIPs)的形成,促進高溶解度和離子電導率。但在連續循環過程中,遊離溶劑分子很容易與電極發生寄生反應。相比之下,弱溶劑化溶劑允許陰離子進入第一溶劑化鞘,有助於接觸離子對(CIP)和離子聚集體(AGGs)的積累,具有更高電化學穩定性,這也增加了陰離子分解機率,陰離子分解主要形成富無機的鈍化產物,這些鈍化產物比傳統低濃度電解質中溶劑分子的富有機分解產物更穩定。然而,過量弱溶劑化溶劑也會導致鹽溶解度和陽離子電導率不足,限制了實際循環性能。此外,CIP/AGG和富無機鈍化可以通過增加鹽與溶劑的比例來形成高濃度電解質(HCEs)。為了提高倍率性能,還可以加入非溶劑化稀釋劑形成局部HCEs,這不僅降低了HCEs的粘度,提高了HCEs的離子電導率,而且保持了CIP/AGG存在。僅用一種溶劑平衡鹽的關聯度(CIP/AGG與SSIP)和溶劑化能力可能具有挑戰性,在Na+體系內更為困難。通過分子設計來調整電解質溶劑的溶劑化能力,使用部分鹵化、甲基化、氰化等來匹配每種鹽的關聯度,是製備穩定性更高的電解質的一種方法,但由於取代基的選擇有限,僅通過分子修飾來優化這些溶劑是很困難的。因此,在這項工作中,作者提出了混合溶劑化電解質(HSEs)策略,包括強溶劑化和弱溶劑化的混合物,它們可以與Na+協同作用,形成分層的溶劑化結構,並相應地實現溶劑化能力的微調,與傳統稀電解質中的強-強溶劑對和LHCE中的強-反溶劑對相比,HSEs具有更大的材料設計空間,可以探索不按年例的強弱溶劑對,弱溶劑化溶劑可以決定氧化還原穩定性和循環極化,而強溶劑化溶劑可以逐漸調節這些性質。這種設計概念也可以擴展到製備鋰金屬電池體系,為電解液設計提供了一個指導原則,使鹼金屬電池的商業化成為可能。
【成果簡介】
近期,美國麻省理工大學李巨教授團隊在Joule上重磅發文:「Hybrid solvating electrolytes for practical sodium-metal batteries」。鈉元素的天然丰度高且成本低廉,鈉金屬電池作為下一代儲能技術顯示出巨大應用潛力,但傳統電解質與高活性鈉金屬負極以及正極的不相容性限制了其性能發揮。本工作報道了一種新型混合溶劑化電解質(HSEs),由鈉鹽的強溶劑和弱溶劑共同組成,實現了電解質關鍵物理和化學性質的調節。研究發現HSEs表現出很強的超混合規則效應,可以在低極化和高氧化還原穩定性之間取得很好的平衡,HSE可以在3.0 mA cm-2的電壓下維持高度可逆的鈉金屬循環,並且能夠匹配高達4.0 V的鈉正極上實現非常穩定的循環性能。這項工作為鈉金屬電池電解液設計提供了一個指導原則,對發展實用化鈉金屬電池成具有重要意義。
【研究內容】
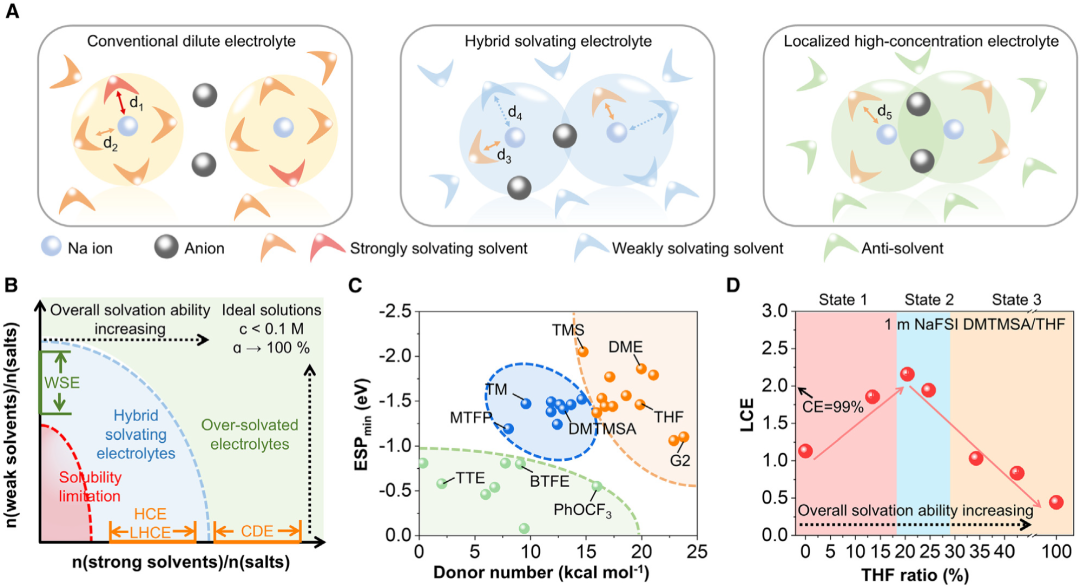 圖1. 混合溶劑化電解質的設計理念
圖1. 混合溶劑化電解質的設計理念HSEs的設計理念。首先,作者根據常見電解液溶劑的供體數(DN)、靜電電位(ESP)以及相對於總可接觸表面的極性面積比等指標對溶劑進行預篩選,這些指標可以有效區分強溶劑化和弱溶劑化溶劑和抗溶劑(圖1)。弱溶劑對可以優化鹽的溶解度和電解質粘度,從而提高陽離子的導電性引起相分離或損害對鈉金屬負極和正極的電化學穩定性。因此,通過快速初始篩選和後續微調,可以快速實現氧化電壓、庫倫效率和過電位等關鍵電化學參數的優化。以DMTMSA和THF進行介紹,因為DMTMSA是弱溶劑溶劑,具有良好的分解產物,THF可以溶解NaFSI,形成7 mol/kg的溶液。通過改變DMTMSA與THF的摩爾比,這些電解質在1 mol/kg NaFSI下的極化曲線相應發生變化。計算了1.0 mA/cm2下100次循環中的平均CEs和循環過電位,以評估它們的電化學性能。結果發現,隨着THF比的增加,平均循環過電位先從> 600 mV下降到72 mV,然後增加到156 mV,平均CE最初從92.60%增加到99.30%,隨後下降到64.02%,這與使用Aurbach方法在1.0 mA/cm2和1.0 mAh/cm2的面容量下觀察到的趨勢相匹配,平均CE和循環過電位之間的非單調相關性指導了HSE設計,當使用NaFSI作為鹽時,DMTMSA和THF的最優摩爾比為4:1。
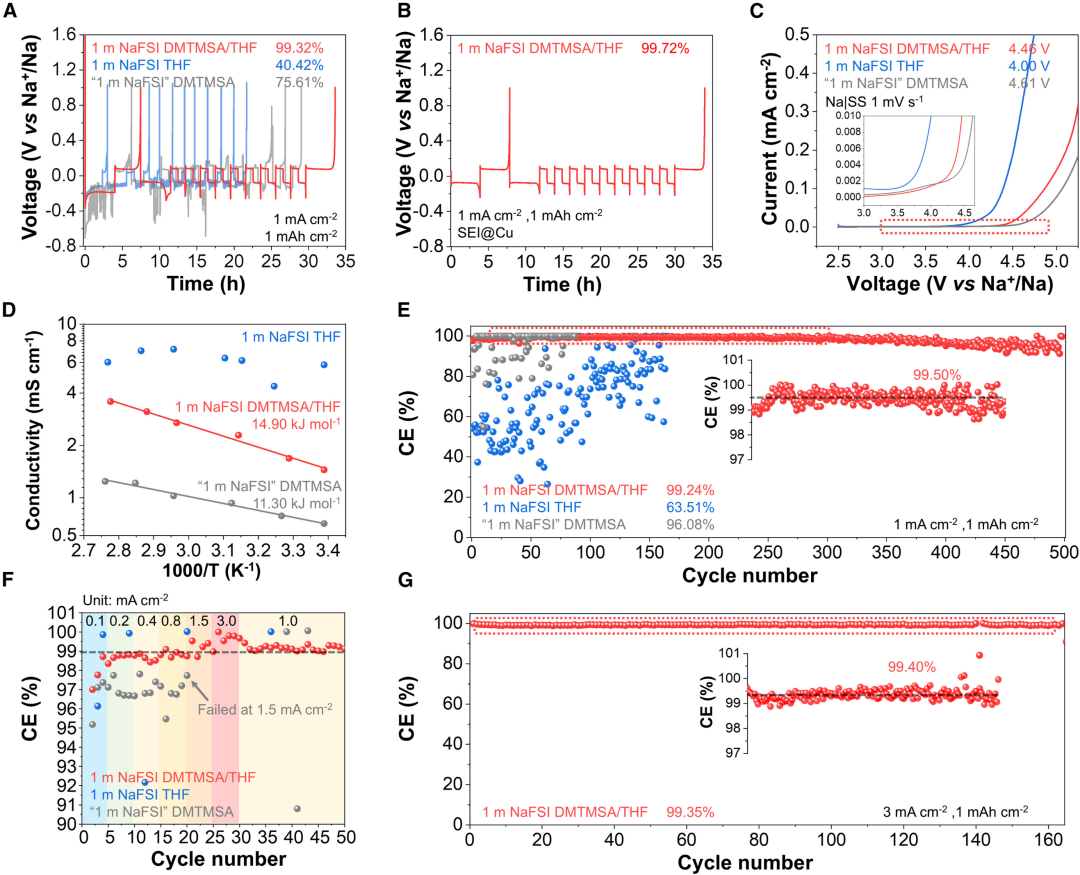 圖2. 雜化溶劑化電解質的電化學評估
圖2. 雜化溶劑化電解質的電化學評估HSEs的電化學評估。為了評價電解質,採用Aurbach方法比較了1.0 mA/cm2下鈉金屬循環的可逆性(圖2)。1 M NaFSI DMTMSA/THF的平均CE達到99.32%(圖2A),這是迄今為止報道的最高記錄,遠優於1 M NaFSI THF(40.42%)和1 M NaFSI DMTMSA(75.61%),優化循環模式後1 M NaFSI DMTMSA/THF的平均CE達到99.72%。使用Na-不鏽鋼(SS)電池進行線性掃描伏安法(LSV),表明,HSE中的THF分子不是「自由」的溶劑分子,而是與Na+協調,增加了氧化穩定性,擴寬了電化學穩定窗口。電化學阻抗譜顯示在室溫下1 M NaFSI DMTMSA/THF的離子電導率比DMTMSA的電解質高220%。1 M NaFSI DMTMSA/THF在室溫下的粘度為5.3 cP,接近類似鹽濃度電解質,觀察到的更高的離子電導率和電化學穩定性是實現快速循環(3.0 mA cm2)和高充電截止電壓(4.0 V)的重要原因。鈉對稱電池測量循環可逆性的方法。1 M NaFSI THF的循環CE波動,而僅使用DMTMSA的電解質的CE波動不太明顯,在100次循環中平均CE為96.1%,後續發生短路。相比之下,1 M NaFSI DMTMSA/THF的初始CE較高,達到95.8%,在前10個循環內CE迅速上升至99.0%,表明該HSE的激活速度很快,在250次循環中,CE保持在99.50%,當電流密度增加到3.0 mA/cm2可以觀察到快速激活,分別經過5次和2次循環後,循環CE超過99.0%。快速活化意味着固體鈍化層的快速形成和氣態和可溶性產物的最小化,這是電解質設計的重要原則之一。在100次循環中,平均CE為99.3%,穩定的循環過電位為80 mV。隨後評估了HSE的倍率性能。對於1 M NaFSI THF,在所有電流密度下都觀察到較差的循環可逆性,而對於僅含DMTMSA的電解質,在電流密度<0.5 mA cm2的情況下,其CE相對較高為97%,循環過電位<100 mV,當電流密度達到1.5 mA cm2時,鍍鈉過程中會出現急劇電壓下降,電位最小值為2.4 V,隨後會發生軟短路。相比之下,1 M NaFSI DMTMSA/THF在3.0 mA cm2下循環穩定,沒有明顯的濃差極化形成,在3.0 mA/cm2下的165次循環後,CE保持在99.4%,並具有穩定的循環過電位。
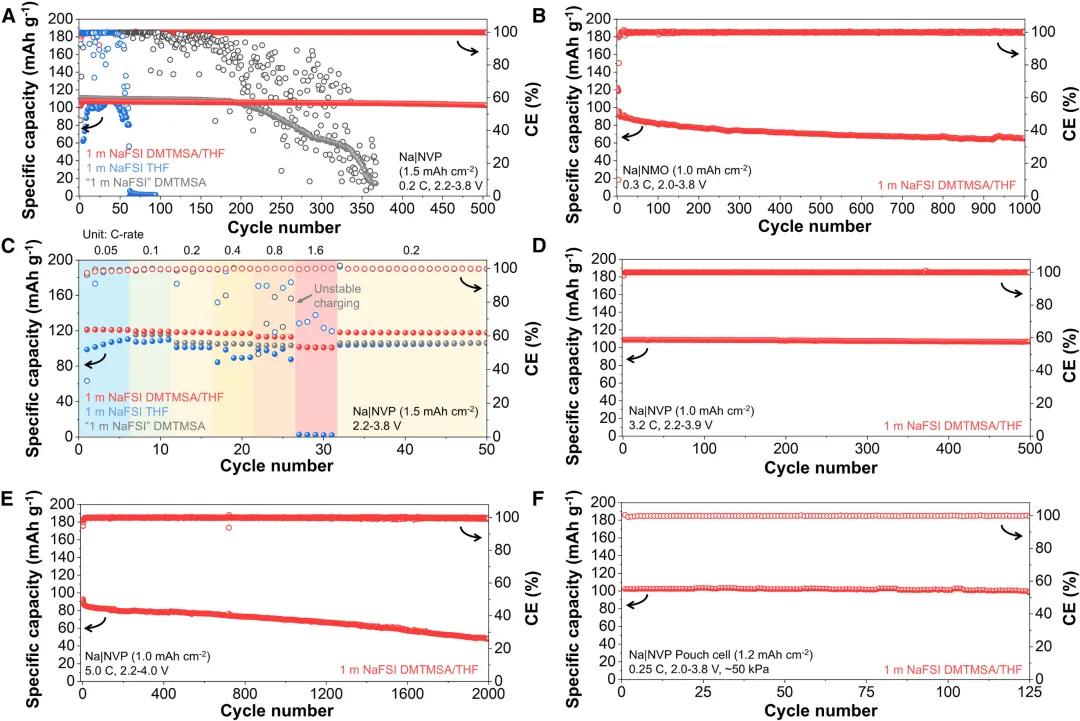 圖3. 雜化溶劑化電解質的電化學穩定性評估
圖3. 雜化溶劑化電解質的電化學穩定性評估HSEs的電化學穩定性評估。高度循環可逆性和氧化穩定性使得該HSE在Na3V2(PO4)3和Na0.44MnO2等常見鈉正極配對時具有廣闊前景。在Na//Na3V2(PO4)3電池中,在0.2 C下,面積容量為1.5 mAh/cm2時,在1 M NaFSI THF下進行50次循環後,觀察到突然的容量損失和極化積累,而使用DMTMSA電解質,由於氧化穩定性的提高,循環壽命可以延長到200次。相比之下,使用1 M NaFSI DMTMSA/THF,循環500次的容量保留率為95.9%,平均CE為99.9%。使用面積容量為1.0 mAh /cm2的Na0.44MnO2正極也可實現類似穩定性,1 M的NaFSI DMTMSA/THF在1000次循環中可以保持71.5%的容量,平均CE為99.9%,優於1 M的NaFSI THF和DMTMSA電解質的平均CE和比容量。此外,使用1 M NaFSI DMTMSA/THF可以觀察到NaNi0.33Fe0.33Mn0.33O2更穩定的循環,而使用1 M NaFSI THF或僅使用DMTMSA的電解質由於氧化不穩定或大極化而無法運行。1 M NaFSI DMTMSA/THF,也可以觀察到倍率性能的改進,在1.6 C下為Na3V2(PO4)3的放電比容量101.7 mAh/g,而THF和DMTMSA的電解質由於充電不穩定和大極化而無法以該倍率循環。當電流密度增加到3.2 C,面積容量為1.0 mAh/cm2,充電截止電壓為3.9 V,使用1 M NaFSI DMTMSA/THF仍可以觀察到穩定的循環,初始比容量為108.9 mAh/g,500個循環後容量保持率為98.1%,平均CE為99.9%。進一步將循環倍率提高到5.0 C,截止電壓提高到4.0 V,初始比容量為92.9 mAh/g,1 M NaFSI DMTMSA/THF在1500個循環中可以保持70%的容量,平均CE為99.9%。以1 M NaFSI DMTMSA/THF電解液組裝的Na//Na3V2(PO4)3軟包電池,經過125次循環後,容量保留為95.8%,平均CE為99.9%,與其他電解液相比表現出優異的可逆性。
 圖4. 雜化溶劑化電解質的微觀和光譜表徵
圖4. 雜化溶劑化電解質的微觀和光譜表徵HSEs的顯微和光譜分析。此外,作者研究了這些不同電解質的Na0沉積形貌(圖4)。對於僅含DMTMSA的電解質,金屬沉積緻密,粒徑較小,平均面積和周長分別為0.43 mm2和3.11mm,高顆粒密度可歸因於高成核密度,僅含DMTMSA的電解質誘導了無機富SEI的形成,可以穩定電極-電解質界面。對於當測試1 M NaFSI THF時,Na0形貌變得鬆散,晶粒尺寸較小,平均晶粒面積為8.7 mm2。相比之下,使用1 M NaFSI DMTMSA/THF,由於穩定的富含無機的SEI和較大的HSE離子電導率,沉積形貌緻密,粒徑較大,SEI的形成是階梯式的,不同元素的還原程度在HSEs中表現出較大的差異。粒子面積與周長之比被定義為結構因子,它與電沉積Na0的尺寸和形狀有關,從1 M NaFSI DMTMSA/THF的結構因子分佈可以看出,大多數微粒顯示出更大值,平均結構因子值分別比1 M NaFSI THF和DMTMSA電解質大4倍和15倍。1 M NaFSI THF的交換電流密度比1 M NaFSI DMTMSA/THF的交換電流密度高2個多數量級。先前研究表明,較高的交換電流密度會引發不太光滑的金屬沉積的形成,這造成CE降低。即使當電流密度增加到3.0 mA/cm2時,使用1 M NaFSI DMTMSA/THF,金屬沉積也會變得緻密,具有大晶粒尺寸。
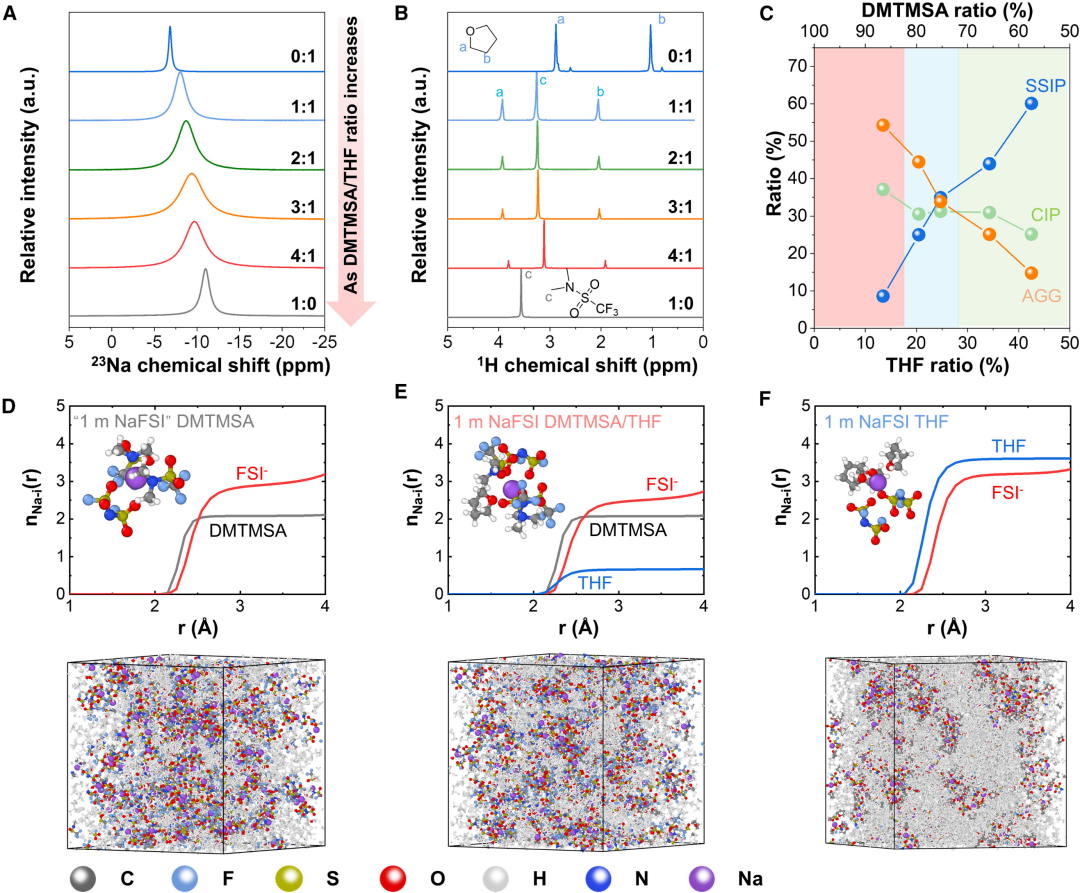 圖5. 雜化溶劑化電解質的性質
圖5. 雜化溶劑化電解質的性質HSEs的溶劑化結構分析。沉積行為優化與HSEs溶劑化結構相關。隨着DMTMSA與THF摩爾比增大,陽離子-偶極相互作用減弱,23Na核磁共振(NMR)峯從6.8 ppm降至11.0 ppm,反映了陽離子附近電子密度增加引起的屏蔽效應,表明陰離子-正離子結合更強,不同HSEs的峯寬均大於單獨THF和DMTMSA電解質。由偶極-偶極相互作用引起的溶劑間相互作用也可以通過THF和DMTMSA的質子化學位移以及DMTMSA的19F NMR化學位移來區分。由於遊離THF的減少和與Na+的配位,THF的質子化學位移隨着DMTMSA/THF比值從0:1增加到1:1而增加。當進一步增加DMTMSA的比值時,可以觀察到輕微下降,這是由溶劑-溶劑相互作用和DMTMSA中富電子O原子在這些質子附近的額外屏蔽效應引起的。拉曼光譜進一步顯示了HSEs中離子-偶極子和偶極子-偶極子相互作用的組合如何調節溶劑化結構。當DMTMSA與THF的摩爾比從1.4增加到6.4時,可以觀察到拉曼峯的藍移。根據計算結果,SSIP從60%下降到10%以下,AGG從14%增加到55%,而CIP從25%到37%略有變化。CIP的形成對於提高電極的熱力學電位是必不可少的,這反過來又削弱了金屬電極的還原能力,從而提高了CE。
為了闡明溶劑化結構並確定Na+溶劑化物在HSEs中的分佈,採用MD模擬計算陰離子與不同溶劑的徑向分佈函數(RDF)和配位結構,由於DMTMSA溶劑的分子結構與FSI-相似,都參與了DMTMSA電解質的Na+溶劑化,DMTMSA和FSI的配位數(CN)分別為2.08和2.86,在初級溶劑化鞘層中主要形成Na+陰離子簇,這是弱溶劑化電解質的典型分子特徵。由於THF與Na+的相互作用比DMTMSA和FSI-更強,部分取代DMTMSA的FSI-可通過控制THF量來實現。此外,在初級溶劑化殼層內,形成了層次化結構,有利於負極附近的逐步脫溶途徑,有助於HSE在循環過程中的低極化。當THF比例從0%增加到20%(階段1)時,可以觀察到THF在初級溶劑化殼內的摻入,AGG和CIP略有下降,這可以維持金屬負極陰離子的優先分解,形成穩定的SEI,同時提高離子電導率。循環過電位從620 mV下降到92 mV,CE從93%上升到99%。對於最佳THF比例的HSE,陰離子CN降低了14%,而THF在初級溶劑化鞘層中降低了13%,進一步提高THF比可將循環過電位降低至72 mV,同時保持相對較高的CE為99%(階段2)。在此階段,可以看到SSIP(35%)、CIP(31%)和AGG(34%)之間的平衡溶劑化結構,其中(AGG + CIP)仍占主導地位。CE的輕微降低可以通過SSIP的增加和THF分子的分解來解釋,隨着THF比的繼續增加,CE降低到64%,過電位增加到156 mV(階段3),這是因為過量的THF會形成過度溶劑化的電解質,其中不協調的溶劑分子占主導地位,由於THF溶劑在正極和負極的不穩定分解,導致CE衰減和極化形成。光譜分析和MD模擬以及電化學性能表明,平衡的溶劑化結構可以是實現快速循環的電解液設計的更好選擇,同時仍然保持對負極和正極的良好穩定性。
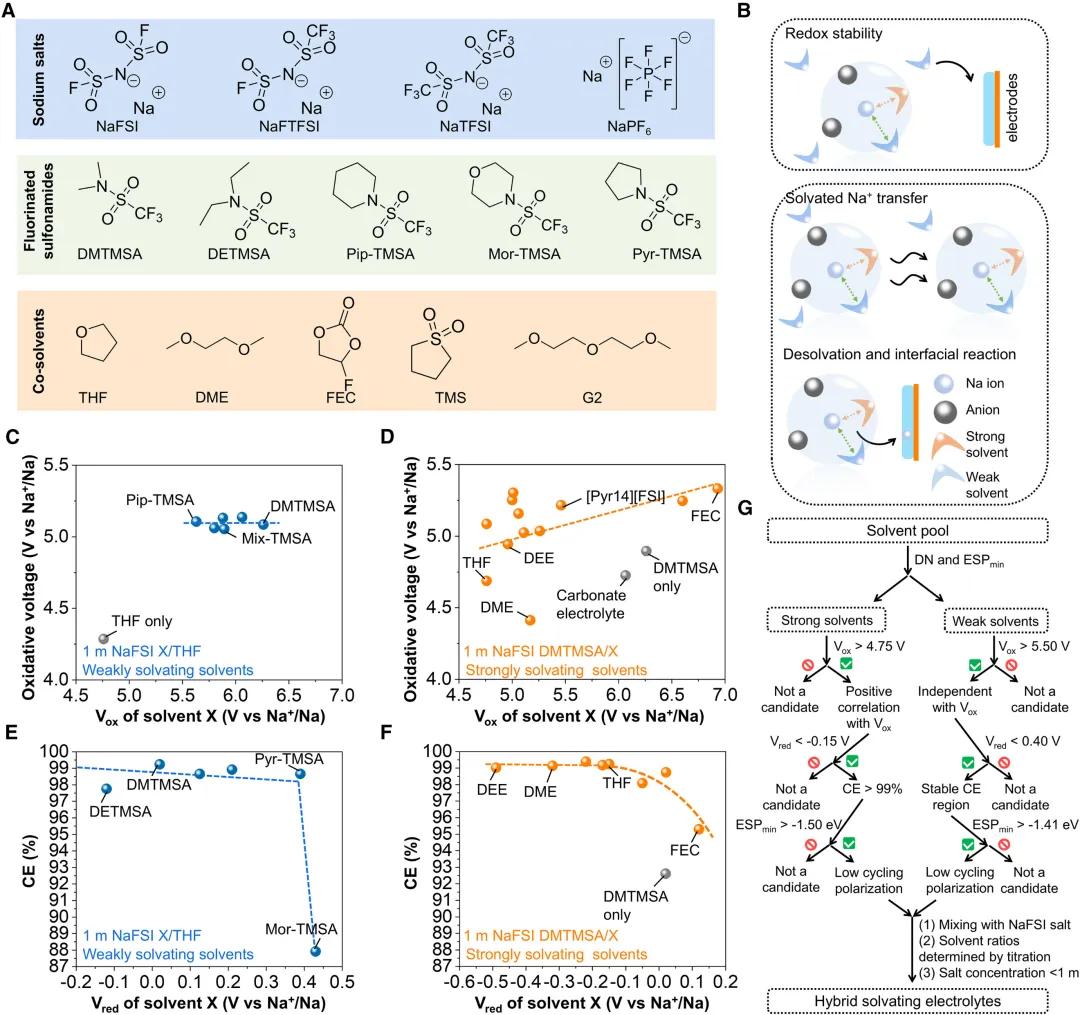 圖6. 混合溶劑化電解質的通用策略
圖6. 混合溶劑化電解質的通用策略設計HSE的通用策略。為了探索不同鈉鹽和溶劑對電極附近氧化還原穩定性、溶劑化Na+移動和脫嵌的影響,作者製備了50種HSEs,分別測試了其的氧化耐受性、對鈉金屬負極的循環穩定性和離子電導率。研究發現,鈉鹽可以分別影響氧化電位和還原電位的氧化穩定性和金屬可逆性,而最負電位的變化對循環過電位的影響較小,隨着鈉鹽氧化電位的增加,可以提高電解質的穩定性,但還原電位與HSEs的平均循環CE呈負相關。因此,為了達到高的平均CE和低循環過電位,與其他鈉鹽相比NaFSI更合適。引入Pip-TMSA使HSE的氧化電壓提高了0.8 V,而用其他弱溶劑替代Pip-TMSA,還原電位高達0.6 V。當使用FEC替代THF時,強溶劑替代可以將氧化電壓提高到5.3 V。在考慮強溶劑和弱溶劑的還原電位和最負電位時,CE和過電位也有類似的趨勢。儘管弱溶劑的溶劑化能力較低,但可以決定HSE的氧化還原穩定性和循環極化,而強溶劑可以進一步微調這些性質。因此,根據溶劑類型的不同,HSE選擇溶劑的標準不同,可以使用溶劑描述符進行預標記。理想的弱溶劑應具有高氧化電壓> 5.50 V和低還原電位 < 0.40 V,而理想的強溶劑應具有高氧化電壓 > 4.75 V和低還原電位 < -0.15 V,以實現高CE > 99%和高氧化電壓> 5.0 V。為了進一步降低循環過電位,建議使用最負ESP > -1.50 eV的強溶劑和最負ESP > -1.41 eV的弱溶劑。結果表明,HSEs的最終鹽濃度為0.8~1.0 mol/kg,強溶劑佔10%~30%。此外,各種弱溶劑和強溶劑的組合可以進一步優化HSEs的電化學性能。未來設計具有更高平均CE(99.8%)和氧化穩定性(與高壓正極配對)的HSEs對於無負極鈉電池在100次循環後實現>80%的高容量保持非常重要。
【文獻總結】
綜上所示,與最近報道鈉金屬電池的先進電解質相比,本工作設計的HSE即使在實際電流密度(3.0 mA cm2)下循環,也能實現最快的激活,並保持低過電位和高可逆性。然而由於缺乏有利的溶劑化結構,這種性能釋放無法通過單一溶劑來實現。通過50個HSE實驗,理清了強溶劑和弱溶劑的選擇原則,以及它們對氧化還原穩定性和循環極化的不同影響。由於高熵電解質可以提高電化學性能,因此多鹽和助溶劑的HSE在未來是一個很有前途的發展方向。此外,HSEs的設計原理也適用於其他可充電電池、液流電池和使用液體電解質的電化學電池,對儲能領域電解液設計具有重要意義。
【文獻信息】
Hybrid solvating electrolytes for practical sodium-metal batteries.
Joule, 2025, 9, 101811.
https://doi.org/10.1016/j.joule.2024.101811

郵發代號:80-732
聯繫熱線:010-64519601/9602/9643
投稿網址:http://esst.cip.com.cn/CN/2095-4239/home.shtml