
【研究背景】
作为电动汽车和电网的下一代储能技术,钠电池展现出巨大的潜力,与锂元素相比,钠元素的天然丰度高、成本更低。钠金属负极具有较高的理论比容量(1165 mAh/g)和低氧化还原电位(2.714 V vs. SHE),将成为是钠电池的终极负极材料。然而,传统电解质与高反应性钠金属负极和正极的不相容性限制了其性能发挥,这些电极的平衡电位在传统电解质的稳定电压窗口之外,导致电解质易发生分解和电极形成不溶性产物。在正极侧,界面副反应也会引起正极颗粒不可逆的表面相变和极化反应,导致容量衰减。在负极侧,在钠金属沉积和剥离过程中,固体电解质界面(SEI)的化学-机械不稳定性导致形态降解和金属分离,加速电解质消耗,降低电池可逆性和循环寿命。此外,枝晶的存在会桥接电极,导致内部短路和热失控。最近,对钠金属电池(SMBs)的研究主要集中在金属钠的电沉积和剥离行为,以及SEI的结构和组成。为了减轻孤立Na0的积累并提高库仑效率(CE),研究者提出了各种策略,包括电解质工程,界面功能化,多孔电极开发,和循环方案优化等。
对于电解质,开发了不同的盐、溶剂、添加剂及其组合,目的是获得更宽的氧化还原电压和循环可逆性。单盐单溶剂电解质需要在低极化和高氧化还原稳定性之间进行权衡。传统电解质使用强阳离子溶剂化溶剂,增强阳离子-偶极相互作用,促进溶剂分离离子对(SSIPs)的形成,促进高溶解度和离子电导率。但在连续循环过程中,游离溶剂分子很容易与电极发生寄生反应。相比之下,弱溶剂化溶剂允许阴离子进入第一溶剂化鞘,有助于接触离子对(CIP)和离子聚集体(AGGs)的积累,具有更高电化学稳定性,这也增加了阴离子分解机率,阴离子分解主要形成富无机的钝化产物,这些钝化产物比传统低浓度电解质中溶剂分子的富有机分解产物更稳定。然而,过量弱溶剂化溶剂也会导致盐溶解度和阳离子电导率不足,限制了实际循环性能。此外,CIP/AGG和富无机钝化可以通过增加盐与溶剂的比例来形成高浓度电解质(HCEs)。为了提高倍率性能,还可以加入非溶剂化稀释剂形成局部HCEs,这不仅降低了HCEs的粘度,提高了HCEs的离子电导率,而且保持了CIP/AGG存在。仅用一种溶剂平衡盐的关联度(CIP/AGG与SSIP)和溶剂化能力可能具有挑战性,在Na+体系内更为困难。通过分子设计来调整电解质溶剂的溶剂化能力,使用部分卤化、甲基化、氰化等来匹配每种盐的关联度,是制备稳定性更高的电解质的一种方法,但由于取代基的选择有限,仅通过分子修饰来优化这些溶剂是很困难的。因此,在这项工作中,作者提出了混合溶剂化电解质(HSEs)策略,包括强溶剂化和弱溶剂化的混合物,它们可以与Na+协同作用,形成分层的溶剂化结构,并相应地实现溶剂化能力的微调,与传统稀电解质中的强-强溶剂对和LHCE中的强-反溶剂对相比,HSEs具有更大的材料设计空间,可以探索不同比例的强弱溶剂对,弱溶剂化溶剂可以决定氧化还原稳定性和循环极化,而强溶剂化溶剂可以逐渐调节这些性质。这种设计概念也可以扩展到制备锂金属电池体系,为电解液设计提供了一个指导原则,使碱金属电池的商业化成为可能。
【成果简介】
近期,美国麻省理工大学李巨教授团队在Joule上重磅发文:“Hybrid solvating electrolytes for practical sodium-metal batteries”。钠元素的天然丰度高且成本低廉,钠金属电池作为下一代储能技术显示出巨大应用潜力,但传统电解质与高活性钠金属负极以及正极的不相容性限制了其性能发挥。本工作报道了一种新型混合溶剂化电解质(HSEs),由钠盐的强溶剂和弱溶剂共同组成,实现了电解质关键物理和化学性质的调节。研究发现HSEs表现出很强的超混合规则效应,可以在低极化和高氧化还原稳定性之间取得很好的平衡,HSE可以在3.0 mA cm-2的电压下维持高度可逆的钠金属循环,并且能够匹配高达4.0 V的钠正极上实现非常稳定的循环性能。这项工作为钠金属电池电解液设计提供了一个指导原则,对发展实用化钠金属电池成具有重要意义。
【研究内容】
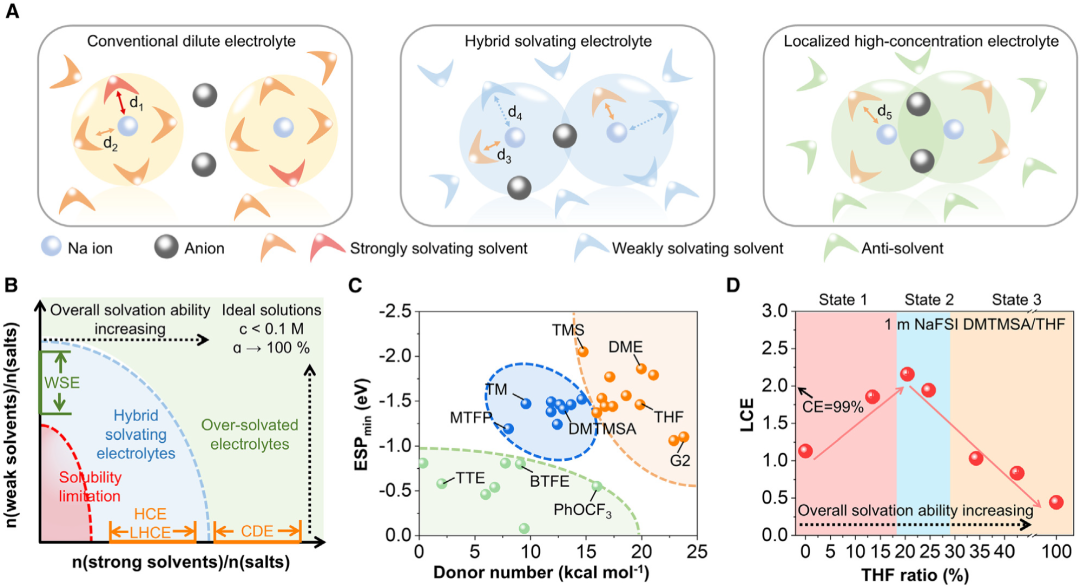 图1. 混合溶剂化电解质的设计理念
图1. 混合溶剂化电解质的设计理念HSEs的设计理念。首先,作者根据常见电解液溶剂的供体数(DN)、静电电位(ESP)以及相对于总可接触表面的极性面积比等指标对溶剂进行预筛选,这些指标可以有效区分强溶剂化和弱溶剂化溶剂和抗溶剂(图1)。弱溶剂对可以优化盐的溶解度和电解质粘度,从而提高阳离子的导电性引起相分离或损害对钠金属负极和正极的电化学稳定性。因此,通过快速初始筛选和后续微调,可以快速实现氧化电压、库伦效率和过电位等关键电化学参数的优化。以DMTMSA和THF进行介绍,因为DMTMSA是弱溶剂溶剂,具有良好的分解产物,THF可以溶解NaFSI,形成7 mol/kg的溶液。通过改变DMTMSA与THF的摩尔比,这些电解质在1 mol/kg NaFSI下的极化曲线相应发生变化。计算了1.0 mA/cm2下100次循环中的平均CEs和循环过电位,以评估它们的电化学性能。结果发现,随着THF比的增加,平均循环过电位先从> 600 mV下降到72 mV,然后增加到156 mV,平均CE最初从92.60%增加到99.30%,随后下降到64.02%,这与使用Aurbach方法在1.0 mA/cm2和1.0 mAh/cm2的面容量下观察到的趋势相匹配,平均CE和循环过电位之间的非单调相关性指导了HSE设计,当使用NaFSI作为盐时,DMTMSA和THF的最优摩尔比为4:1。
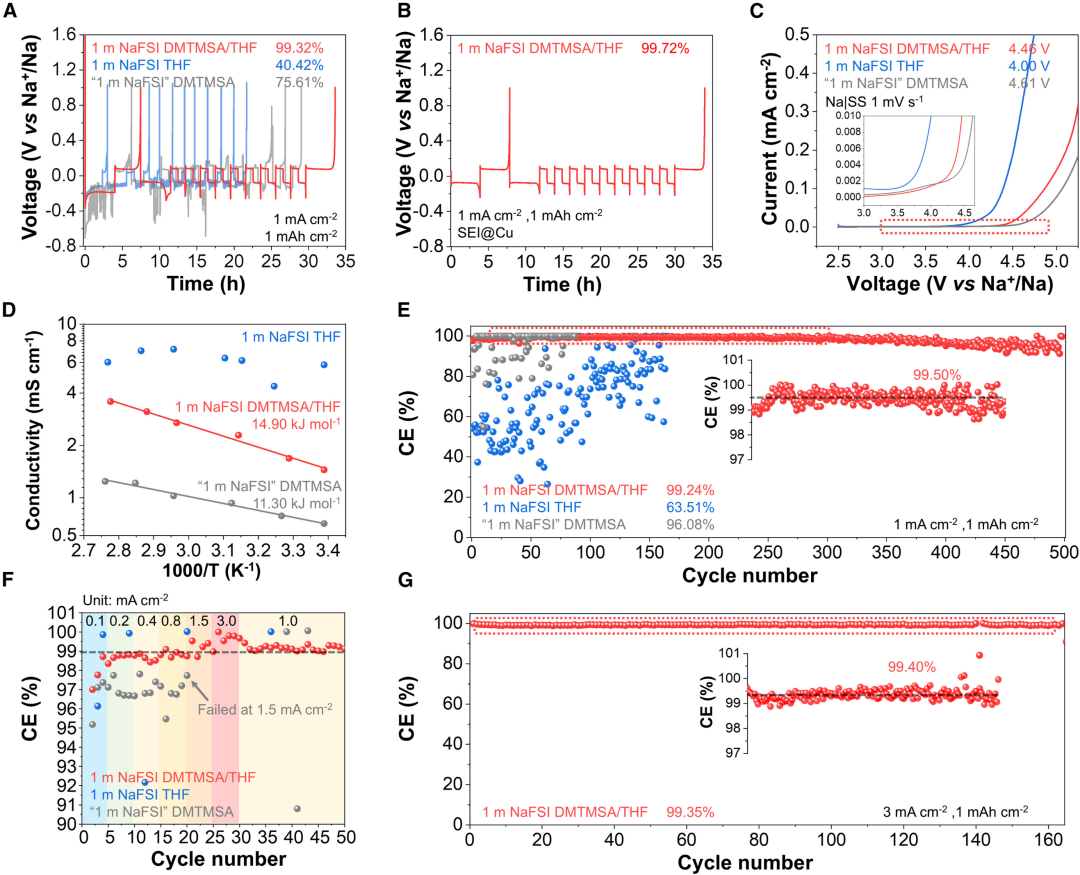 图2. 杂化溶剂化电解质的电化学评估
图2. 杂化溶剂化电解质的电化学评估HSEs的电化学评估。为了评价电解质,采用Aurbach方法比较了1.0 mA/cm2下钠金属循环的可逆性(图2)。1 M NaFSI DMTMSA/THF的平均CE达到99.32%(图2A),这是迄今为止报道的最高记录,远优于1 M NaFSI THF(40.42%)和1 M NaFSI DMTMSA(75.61%),优化循环模式后1 M NaFSI DMTMSA/THF的平均CE达到99.72%。使用Na-不锈钢(SS)电池进行线性扫描伏安法(LSV),表明,HSE中的THF分子不是“自由”的溶剂分子,而是与Na+协调,增加了氧化稳定性,扩宽了电化学稳定窗口。电化学阻抗谱显示在室温下1 M NaFSI DMTMSA/THF的离子电导率比DMTMSA的电解质高220%。1 M NaFSI DMTMSA/THF在室温下的粘度为5.3 cP,接近类似盐浓度电解质,观察到的更高的离子电导率和电化学稳定性是实现快速循环(3.0 mA cm2)和高充电截止电压(4.0 V)的重要原因。钠对称电池测量循环可逆性的方法。1 M NaFSI THF的循环CE波动,而仅使用DMTMSA的电解质的CE波动不太明显,在100次循环中平均CE为96.1%,后续发生短路。相比之下,1 M NaFSI DMTMSA/THF的初始CE较高,达到95.8%,在前10个循环内CE迅速上升至99.0%,表明该HSE的激活速度很快,在250次循环中,CE保持在99.50%,当电流密度增加到3.0 mA/cm2可以观察到快速激活,分别经过5次和2次循环后,循环CE超过99.0%。快速活化意味着固体钝化层的快速形成和气态和可溶性产物的最小化,这是电解质设计的重要原则之一。在100次循环中,平均CE为99.3%,稳定的循环过电位为80 mV。随后评估了HSE的倍率性能。对于1 M NaFSI THF,在所有电流密度下都观察到较差的循环可逆性,而对于仅含DMTMSA的电解质,在电流密度<0.5 mA cm2的情况下,其CE相对较高为97%,循环过电位<100 mV,当电流密度达到1.5 mA cm2时,镀钠过程中会出现急剧电压下降,电位最小值为2.4 V,随后会发生软短路。相比之下,1 M NaFSI DMTMSA/THF在3.0 mA cm2下循环稳定,没有明显的浓差极化形成,在3.0 mA/cm2下的165次循环后,CE保持在99.4%,并具有稳定的循环过电位。
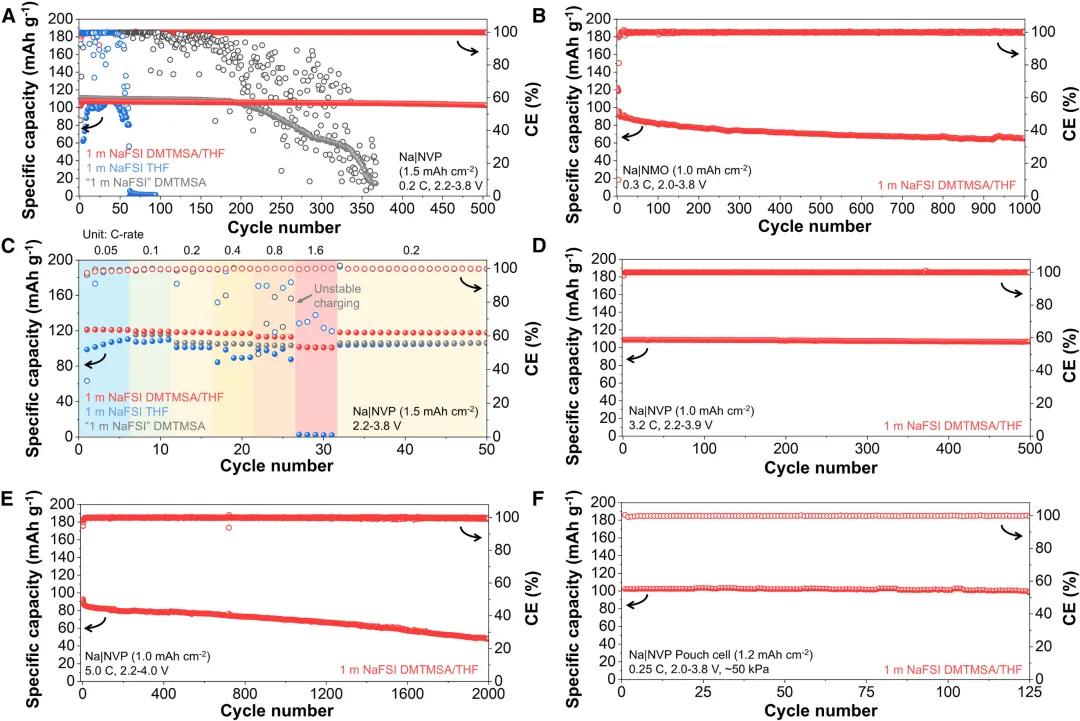 图3. 杂化溶剂化电解质的电化学稳定性评估
图3. 杂化溶剂化电解质的电化学稳定性评估HSEs的电化学稳定性评估。高度循环可逆性和氧化稳定性使得该HSE在Na3V2(PO4)3和Na0.44MnO2等常见钠正极配对时具有广阔前景。在Na//Na3V2(PO4)3电池中,在0.2 C下,面积容量为1.5 mAh/cm2时,在1 M NaFSI THF下进行50次循环后,观察到突然的容量损失和极化积累,而使用DMTMSA电解质,由于氧化稳定性的提高,循环寿命可以延长到200次。相比之下,使用1 M NaFSI DMTMSA/THF,循环500次的容量保留率为95.9%,平均CE为99.9%。使用面积容量为1.0 mAh /cm2的Na0.44MnO2正极也可实现类似稳定性,1 M的NaFSI DMTMSA/THF在1000次循环中可以保持71.5%的容量,平均CE为99.9%,优于1 M的NaFSI THF和DMTMSA电解质的平均CE和比容量。此外,使用1 M NaFSI DMTMSA/THF可以观察到NaNi0.33Fe0.33Mn0.33O2更稳定的循环,而使用1 M NaFSI THF或仅使用DMTMSA的电解质由于氧化不稳定或大极化而无法运行。1 M NaFSI DMTMSA/THF,也可以观察到倍率性能的改进,在1.6 C下为Na3V2(PO4)3的放电比容量101.7 mAh/g,而THF和DMTMSA的电解质由于充电不稳定和大极化而无法以该倍率循环。当电流密度增加到3.2 C,面积容量为1.0 mAh/cm2,充电截止电压为3.9 V,使用1 M NaFSI DMTMSA/THF仍可以观察到稳定的循环,初始比容量为108.9 mAh/g,500个循环后容量保持率为98.1%,平均CE为99.9%。进一步将循环倍率提高到5.0 C,截止电压提高到4.0 V,初始比容量为92.9 mAh/g,1 M NaFSI DMTMSA/THF在1500个循环中可以保持70%的容量,平均CE为99.9%。以1 M NaFSI DMTMSA/THF电解液组装的Na//Na3V2(PO4)3软包电池,经过125次循环后,容量保留为95.8%,平均CE为99.9%,与其他电解液相比表现出优异的可逆性。
 图4. 杂化溶剂化电解质的微观和光谱表征
图4. 杂化溶剂化电解质的微观和光谱表征HSEs的显微和光谱分析。此外,作者研究了这些不同电解质的Na0沉积形貌(图4)。对于仅含DMTMSA的电解质,金属沉积致密,粒径较小,平均面积和周长分别为0.43 mm2和3.11mm,高颗粒密度可归因于高成核密度,仅含DMTMSA的电解质诱导了无机富SEI的形成,可以稳定电极-电解质界面。对于当测试1 M NaFSI THF时,Na0形貌变得松散,晶粒尺寸较小,平均晶粒面积为8.7 mm2。相比之下,使用1 M NaFSI DMTMSA/THF,由于稳定的富含无机的SEI和较大的HSE离子电导率,沉积形貌致密,粒径较大,SEI的形成是阶梯式的,不同元素的还原程度在HSEs中表现出较大的差异。粒子面积与周长之比被定义为结构因子,它与电沉积Na0的尺寸和形状有关,从1 M NaFSI DMTMSA/THF的结构因子分布可以看出,大多数微粒显示出更大值,平均结构因子值分别比1 M NaFSI THF和DMTMSA电解质大4倍和15倍。1 M NaFSI THF的交换电流密度比1 M NaFSI DMTMSA/THF的交换电流密度高2个多数量级。先前研究表明,较高的交换电流密度会引发不太光滑的金属沉积的形成,这造成CE降低。即使当电流密度增加到3.0 mA/cm2时,使用1 M NaFSI DMTMSA/THF,金属沉积也会变得致密,具有大晶粒尺寸。
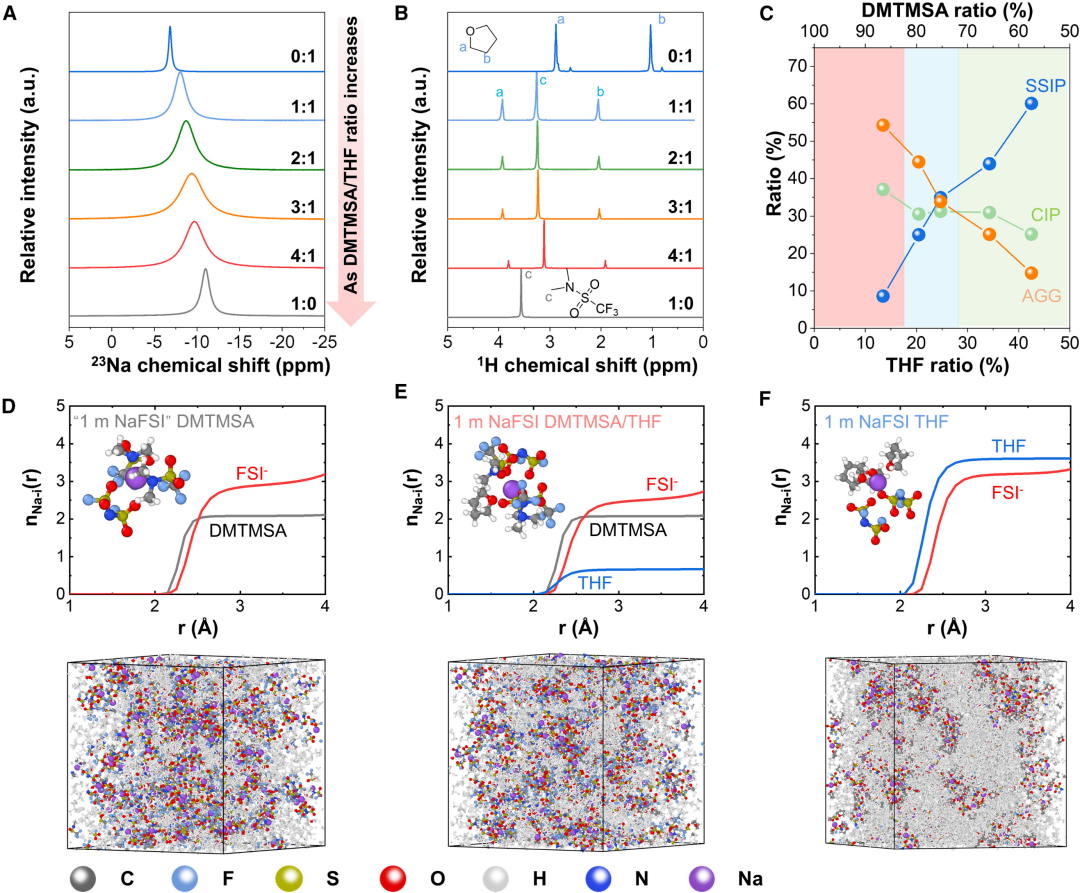 图5. 杂化溶剂化电解质的性质
图5. 杂化溶剂化电解质的性质HSEs的溶剂化结构分析。沉积行为优化与HSEs溶剂化结构相关。随着DMTMSA与THF摩尔比增大,阳离子-偶极相互作用减弱,23Na核磁共振(NMR)峰从6.8 ppm降至11.0 ppm,反映了阳离子附近电子密度增加引起的屏蔽效应,表明阴离子-正离子结合更强,不同HSEs的峰宽均大于单独THF和DMTMSA电解质。由偶极-偶极相互作用引起的溶剂间相互作用也可以通过THF和DMTMSA的质子化学位移以及DMTMSA的19F NMR化学位移来区分。由于游离THF的减少和与Na+的配位,THF的质子化学位移随着DMTMSA/THF比值从0:1增加到1:1而增加。当进一步增加DMTMSA的比值时,可以观察到轻微下降,这是由溶剂-溶剂相互作用和DMTMSA中富电子O原子在这些质子附近的额外屏蔽效应引起的。拉曼光谱进一步显示了HSEs中离子-偶极子和偶极子-偶极子相互作用的组合如何调节溶剂化结构。当DMTMSA与THF的摩尔比从1.4增加到6.4时,可以观察到拉曼峰的蓝移。根据计算结果,SSIP从60%下降到10%以下,AGG从14%增加到55%,而CIP从25%到37%略有变化。CIP的形成对于提高电极的热力学电位是必不可少的,这反过来又削弱了金属电极的还原能力,从而提高了CE。
为了阐明溶剂化结构并确定Na+溶剂化物在HSEs中的分布,采用MD模拟计算阴离子与不同溶剂的径向分布函数(RDF)和配位结构,由于DMTMSA溶剂的分子结构与FSI-相似,都参与了DMTMSA电解质的Na+溶剂化,DMTMSA和FSI的配位数(CN)分别为2.08和2.86,在初级溶剂化鞘层中主要形成Na+阴离子簇,这是弱溶剂化电解质的典型分子特征。由于THF与Na+的相互作用比DMTMSA和FSI-更强,部分取代DMTMSA的FSI-可通过控制THF量来实现。此外,在初级溶剂化壳层内,形成了层次化结构,有利于负极附近的逐步脱溶途径,有助于HSE在循环过程中的低极化。当THF比例从0%增加到20%(阶段1)时,可以观察到THF在初级溶剂化壳内的掺入,AGG和CIP略有下降,这可以维持金属负极阴离子的优先分解,形成稳定的SEI,同时提高离子电导率。循环过电位从620 mV下降到92 mV,CE从93%上升到99%。对于最佳THF比例的HSE,阴离子CN降低了14%,而THF在初级溶剂化鞘层中降低了13%,进一步提高THF比可将循环过电位降低至72 mV,同时保持相对较高的CE为99%(阶段2)。在此阶段,可以看到SSIP(35%)、CIP(31%)和AGG(34%)之间的平衡溶剂化结构,其中(AGG + CIP)仍占主导地位。CE的轻微降低可以通过SSIP的增加和THF分子的分解来解释,随着THF比的继续增加,CE降低到64%,过电位增加到156 mV(阶段3),这是因为过量的THF会形成过度溶剂化的电解质,其中不协调的溶剂分子占主导地位,由于THF溶剂在正极和负极的不稳定分解,导致CE衰减和极化形成。光谱分析和MD模拟以及电化学性能表明,平衡的溶剂化结构可以是实现快速循环的电解液设计的更好选择,同时仍然保持对负极和正极的良好稳定性。
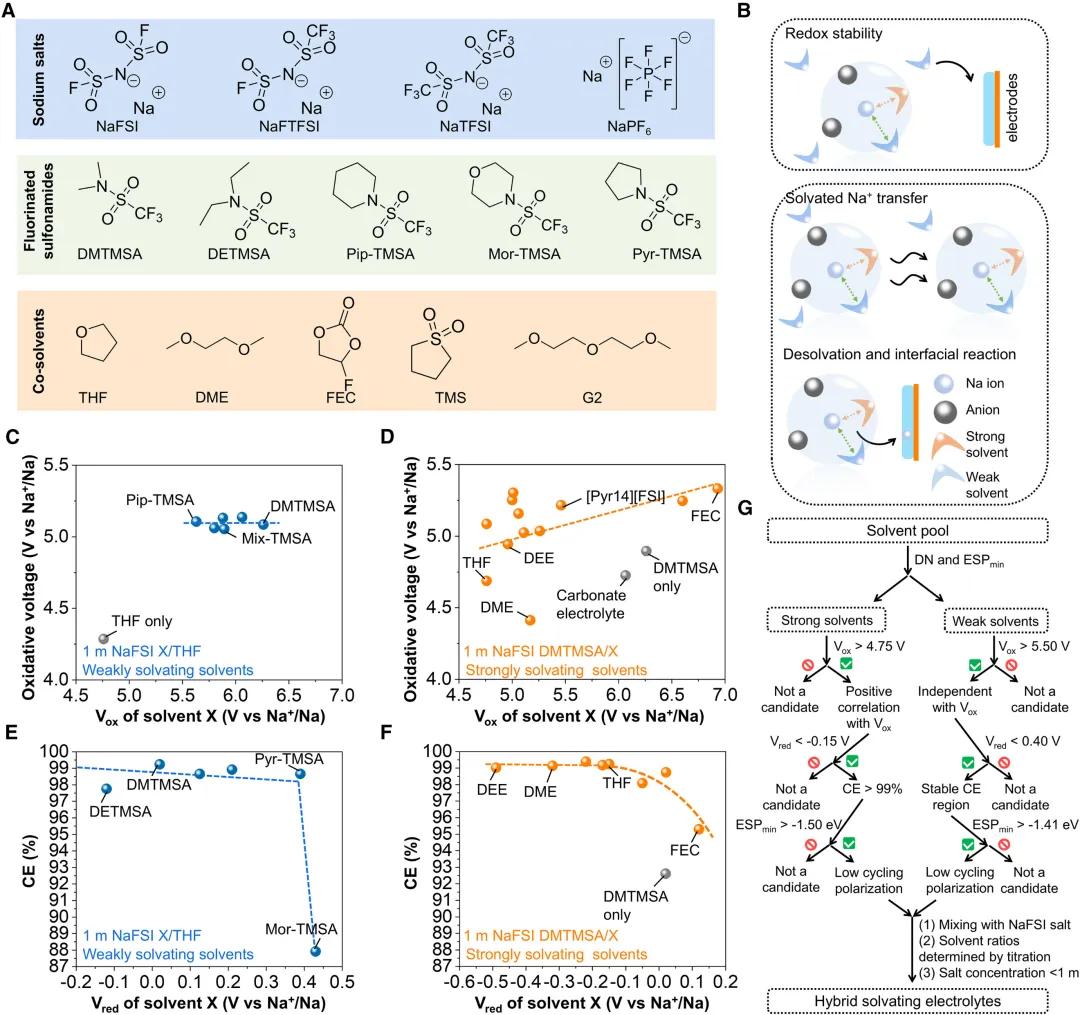 图6. 混合溶剂化电解质的通用策略
图6. 混合溶剂化电解质的通用策略设计HSE的通用策略。为了探索不同钠盐和溶剂对电极附近氧化还原稳定性、溶剂化Na+移动和脱嵌的影响,作者制备了50种HSEs,分别测试了其的氧化耐受性、对钠金属负极的循环稳定性和离子电导率。研究发现,钠盐可以分别影响氧化电位和还原电位的氧化稳定性和金属可逆性,而最负电位的变化对循环过电位的影响较小,随着钠盐氧化电位的增加,可以提高电解质的稳定性,但还原电位与HSEs的平均循环CE呈负相关。因此,为了达到高的平均CE和低循环过电位,与其他钠盐相比NaFSI更合适。引入Pip-TMSA使HSE的氧化电压提高了0.8 V,而用其他弱溶剂替代Pip-TMSA,还原电位高达0.6 V。当使用FEC替代THF时,强溶剂替代可以将氧化电压提高到5.3 V。在考虑强溶剂和弱溶剂的还原电位和最负电位时,CE和过电位也有类似的趋势。尽管弱溶剂的溶剂化能力较低,但可以决定HSE的氧化还原稳定性和循环极化,而强溶剂可以进一步微调这些性质。因此,根据溶剂类型的不同,HSE选择溶剂的标准不同,可以使用溶剂描述符进行预标记。理想的弱溶剂应具有高氧化电压> 5.50 V和低还原电位 < 0.40 V,而理想的强溶剂应具有高氧化电压 > 4.75 V和低还原电位 < -0.15 V,以实现高CE > 99%和高氧化电压> 5.0 V。为了进一步降低循环过电位,建议使用最负ESP > -1.50 eV的强溶剂和最负ESP > -1.41 eV的弱溶剂。结果表明,HSEs的最终盐浓度为0.8~1.0 mol/kg,强溶剂占10%~30%。此外,各种弱溶剂和强溶剂的组合可以进一步优化HSEs的电化学性能。未来设计具有更高平均CE(99.8%)和氧化稳定性(与高压正极配对)的HSEs对于无负极钠电池在100次循环后实现>80%的高容量保持非常重要。
【文献总结】
综上所示,与最近报道钠金属电池的先进电解质相比,本工作设计的HSE即使在实际电流密度(3.0 mA cm2)下循环,也能实现最快的激活,并保持低过电位和高可逆性。然而由于缺乏有利的溶剂化结构,这种性能释放无法通过单一溶剂来实现。通过50个HSE实验,理清了强溶剂和弱溶剂的选择原则,以及它们对氧化还原稳定性和循环极化的不同影响。由于高熵电解质可以提高电化学性能,因此多盐和助溶剂的HSE在未来是一个很有前途的发展方向。此外,HSEs的设计原理也适用于其他可充电电池、液流电池和使用液体电解质的电化学电池,对储能领域电解液设计具有重要意义。
【文献信息】
Hybrid solvating electrolytes for practical sodium-metal batteries.
Joule, 2025, 9, 101811.
https://doi.org/10.1016/j.joule.2024.101811

邮发代号:80-732
联系热线:010-64519601/9602/9643
投稿网址:http://esst.cip.com.cn/CN/2095-4239/home.shtml